| 1.0 | OBJECTIVE:
To lay down a procedure for investigation and evaluation of out-of-specification test results obtained during laboratory testing of drug products, drug substances, intermediates, raw materials and packaging materials. |
|||||||||||||||||||||
| 2.0 | SCOPE:
Applicable to
Not Applicable to: This procedure may not be applicable for Development/Engineering batches, Analyst Qualification, Cleaning validation, analysis of innovator sample, Market complain sample which are not stored at defined storage conditions. |
|||||||||||||||||||||
| 3.0 | RESPONSIBILITY: | |||||||||||||||||||||
| 3.1 | QC Analyst: To report the OOS result, retain all the test solutions and glassware for investigation, participate in retesting and investigation. | |||||||||||||||||||||
| 3.2 | QC Section in-charge: To review the analytical data and OOS result, issue investigation form, immediately inform to QA, Check list based investigation, interview of analyst, lab investigation and plan for retest. | |||||||||||||||||||||
| 3.3 | Production in-charge: To carry out manufacturing investigation. | |||||||||||||||||||||
| 3.4 | QA Team: Log OOS, assign the OOS reference number, review the OOS test result, plan for full scale investigation and prepare a final investigation report. Involve in investigation and take a final decision on batch disposition. | |||||||||||||||||||||
| 3.5 | Process Development / R&D in-charge: To provide support in investigation. | |||||||||||||||||||||
| 3.6 | QA Head: Inform to regulatory authority for commercial distributed batch / product failure. Approval of batch disposition and ensure closure of CAPA. | |||||||||||||||||||||
| 4.0 | DEFINITIONS: | |||||||||||||||||||||
| 4.1 | Out-Of-Specification (OOS) Test Result:
A test result that falls outside the established specification or acceptance criteria. |
|||||||||||||||||||||
| 4.2 | Laboratory error:
An error associated with the performance of a test procedure or due to laboratory equipment malfunction or failure. |
|||||||||||||||||||||
| 4.3 | Retesting:
Retesting is –
|
|||||||||||||||||||||
| 4.4 | Resample:
A second or additional sample collected from a lot or a batch of drug substance or drug product by following a standard sampling procedure. |
|||||||||||||||||||||
| 4.5 | Probable cause:
Cause which is likely to have resulted in an out-of-specification test result, but has not been scientifically proven. |
|||||||||||||||||||||
| 4.6 | Obvious Cause:
Cause which is identified during preliminary investigation (check list based investigation) e.g. Calculation error, wrong volumetric flask (100 ml instead of 200 ml) etc. Assignable cause: A scientifically justified explanation of the reason for an out-of-specification test result identified in hypothesis testing and documented during investigation. |
|||||||||||||||||||||
| 4.7 | Batch Disposition:
Action to be taken on the batch in question. (Approve / Reject / Destruction / Reprocessing / Reworking). |
|||||||||||||||||||||
| 4.8 | Laboratory Investigation (Phase I)
Investigation carried out to assess the accuracy of laboratory testing and recording in order to identify whether there was any error in testing of the batch in question that could have resulted in the OOS result. This investigation includes review of sample collection, storage, preparation and testing in the laboratory against the documented procedures. It may also include additional testing. |
|||||||||||||||||||||
| 4.9 | Full scale OOS investigation (Phase II):
Review of manufacturing process and records and additional testing in the laboratory, where required to identify or confirm the cause. |
|||||||||||||||||||||
| 4.10 | Hypothesis Testing:
Testing conducted at laboratory to verify the possible laboratory errors through hypothesis regarding what might have happened (e.g. instrument malfunction, dilution error, incomplete extraction, etc.) |
|||||||||||||||||||||
| 4.11 | Simulation Testing:
Testing conducted at laboratory to identify probable errors by recreating the analysis condition that can result in sample failure. |
|||||||||||||||||||||
| 5.0 | PROCEDURE FOR DRUG SUBSTANCES, INTERMEDIATES, IN-PROCESS MATERIALS AND DRUG PRODUCTS: | |||||||||||||||||||||
| 5.1 | Phase I – Laboratory Investigation | |||||||||||||||||||||
| 5.1.1 | Any OOS test result obtained shall be immediately reported by QC analyst to the Section In-charge. | |||||||||||||||||||||
| 5.1.2 | The analyst shall retain all test preparations, glassware used, portion of the sample solution, standard solution, chromatograms, Test Data Sheets (TDS), any records, etc. for investigation. | |||||||||||||||||||||
| 5.1.3 | Section In-charge shall issue an OOS investigation, Annexure 1, to the analyst who shall record all relevant details in Section A of the form. | |||||||||||||||||||||
| 5.1.4 | Section In-charge shall send the OOS investigation form to QA; QA shall enter the details in the ‘Log for OOS Test Results’, Annexure 2 and assign a unique OOS Report Number representing the serial no. for a particular year. The same number shall also be entered in the investigation form. | |||||||||||||||||||||
| The OOS Investigation Form shall be assigned a Report No. in the form of OOS/AA/BB/XXX, Where,
OOS : Out of Specification. AA : Abbreviation for nature of sample for which OOS is found e.g. AP : Drug Substances, Intermediates FP : Drugs Products RM: Raw Material PM: Packing Material ST: Stability Testing Samples BB : The last two digits of the year in which OOS is found. XXX : The serial number of OOS in a year (i.e. Jan – Dec) |
||||||||||||||||||||||
| After entering the serial no. of OOS, QA person shall check whether similar OOS was reported earlier for same product and also review the outcome of that investigation in light of the current investigation for any recurring error. Same shall be mentioned in the investigation form. | ||||||||||||||||||||||
| 5.1.5 | Section In-charge, along with the analyst, shall investigate the OOS test result as per procedure given below and record the findings in the Section B of the ‘OOS Investigation Form’, | |||||||||||||||||||||
| 5.1.6 | Section In-charge shall –
Glassware used, equipment usage logs, instrument calibration records, solution preparation records, RS/WS usage , chromatograms, calculations, qualification of analyst, trend data for past batches, method validation data wherever applicable etc. (Note- the list is not exhaustive and review may be extended to other relevant documents). Note: Solution preparations older than 2 days of preparation shall be discarded after verification and shall not be considered for further investigational analysis.
|
|||||||||||||||||||||
| 5.1.7 | During investigation, if any lab error is identified as assignable cause, it shall be corrected as per the method of correction recommended in Annexure 3. Any retesting shall be authorized by QC In-charge as per the steps described below-
Table – 1
|
|||||||||||||||||||||
| 5.1.8 | If no lab error is identified as assignable cause during laboratory investigation, simulation test may be conducted to identify the probable errors. | |||||||||||||||||||||
| 5.1.9 | A documented protocol for simulation shall be prepared by QC In-charge and approved by QA In-charge. Objective of this exercise shall be clearly documented in the protocol. Simulation test may include, but not limited to – testing for dilution error, sonication error, heating error, incomplete extraction etc. The outcome of the simulation shall be documented and conclusion shall be drawn. The raw data for the simulation shall be retained with OOS documentation. | |||||||||||||||||||||
| 5.1.10 | If laboratory error is identified in simulation test, it shall be handled as per step 5.1.7. If no obvious laboratory error is identified, report the probable errors (if any) and proceed for further investigation. | |||||||||||||||||||||
| 5.1.11 | If an obvious sampling error or improper handling / storage of sample is detected during investigation, a re-sampling shall be approved by QA. This approval shall be documented . Steps described below shall be undertaken in this case –
|
|||||||||||||||||||||
| 5.1.12 | In the case of microbiological assays if the assignable cause for OOS is not associated with laboratory error, it shall be dealt as per the procedure described in the respective pharmacopoeia.
If assignable cause is identified, the test shall be repeated by following steps ‘a’ to ‘f’ given in the 5.1.7. |
|||||||||||||||||||||
| 5.1.13 | In case of tests which involve multi stage testing as per respective Pharmacopoeia or site SOP, such as Uniformity of Dosage unit, Dissolution, blend uniformity etc.; if the sample does not comply with first stage acceptance criteria, then preliminary laboratory investigation shall be carried out to evaluate any laboratory error.
If there is any laboratory error identified in preliminary investigation, then OOS shall be logged and further investigation shall be carried out. If there is no laboratory error identified in preliminary investigation, then additional testing stages shall be completed as per respective Pharmacopoeia or site SOP. In such cases OOS shall be logged if sample fails to meet the Acceptance criteria after the final stage. |
|||||||||||||||||||||
| 5.1.14 | In case of OOS result for qualitative tests such as description, appearance etc. shall be investigated to determine the cause; extensive re-inspection of the batch(es) shall be performed and fresh sample shall be drawn to conduct any further investigations. This re-sampling shall be approved by QA. | |||||||||||||||||||||
| 5.1.15 | If investigation reveals that the initial sampling method was faulty, a revised correct sampling method shall be developed and documented. This shall be reviewed and approved by QA in-charge. Steps described below shall be undertaken in this case –
|
|||||||||||||||||||||
| 5.1.16 | Laboratory investigation (including preliminary investigation, hypothesis testing, simulation testing etc.) shall be completed within 10 days. In case the laboratory investigation requires more than 10 days, the same shall be justified by the QC In-charge in ‘OOS Investigation form’ (Annexure-1). | |||||||||||||||||||||
| 5.1.17 | If no cause is detected during any of the laboratory investigation, the OOS result stands valid, however the investigation shall be deemed as ‘inconclusive’. In-charge of QC shall state details in the investigation form and submit the same to In-charge of QA. In-charge of QA shall then initiate a full-scale investigation. | |||||||||||||||||||||
| 5.2 | Phase II – Full Scale OOS Investigation
Full scale investigation of the OOS result shall be conducted jointly by QA and production to identify error related to manufacturing and packing process. Other departments like Process Development, R&D and engineering may be involved. The investigation shall include as applicable, but not restricted to: |
|||||||||||||||||||||
| 5.2.1 | Review Of Production Process And Records | |||||||||||||||||||||
| 5.2.1.1 | On receiving the information for initiation of OOS, primary manufacturing investigation related to review of production process and records shall be started. The investigation shall include but shall not be restricted to (a) Review of Input material trends; (b) Batch Record – to check any deviation from manufacturing procedure or equipment operation procedure or environment monitoring parameter, and yield at different stages, etc.; (c) Trend data for past batches; (d) Equipment maintenance records; (e) Changes in facility / equipment / process; (f) Environment monitoring results. Record the investigation in the form ‘Manufacturing Investigation Form’ (Annexure 4). | |||||||||||||||||||||
| 5.2.1.2 | During review, if any assignable cause that confirms the OOS result is identified, the OOS investigation shall be terminated and decision on the disposition of the batch shall be decided by QA In-charge. A failure investigation shall be initiated. The investigation shall be extended to other batches of the same product / different product, where applicable. | |||||||||||||||||||||
| 5.2.1.3 | Primary manufacturing investigation shall be completed within 7 days from the initiation of OOS. In case the investigation requires more than 7 days, the same shall be justified by the Production In-charge in ‘Manufacturing Investigation Form’ (Annexure 4). | |||||||||||||||||||||
| 5.2.1.4 | For unstable intermediates, as per Annexure 8, the primary manufacturing investigation shall be completed as mentioned in Pt. 5.2.1.3. Meanwhile, the OOS batch shall be preceded further, without waiting for the primary manufacturing investigation to complete, till a stable process stage is achieved. | |||||||||||||||||||||
| 5.2.2 | Experimental Investigation | |||||||||||||||||||||
| 5.2.2.1 | Where necessary, hypothesis about probable error in manufacturing process shall be verified by Process development lab / R & D through lab experiments by simulating the error. This shall be done against a documented protocol that is approved by QA. The protocol shall clearly describe the objective of the experiment. | |||||||||||||||||||||
| 5.2.2.2 | A documented protocol for this exercise shall be prepared by Production / PDL In-charge and approved by QA In-charge. Objective of this exercise shall be clearly documented in the protocol. The outcome of the hypothesis shall be documented and conclusion shall be drawn. | |||||||||||||||||||||
| 5.2.2.3 | If no manufacturing error is observed, retesting shall be conducted to rule out possibility of OOS result due to any unidentified laboratory error. | |||||||||||||||||||||
| 5.2.3 | Retesting: | |||||||||||||||||||||
| 5.2.3.1 | Retesting may be initiated in parallel with manufacturing investigation to save time; however decision on the batch in such case shall be taken only after completion of manufacturing investigation. | |||||||||||||||||||||
| 5.2.3.2 | In case, if retesting in triplicate is done during laboratory investigations for any observed / identified lab error and proceeded further for full scale investigation; further retesting shall not be done and decision for the disposition of the batch shall be taken by QA In-charge. | |||||||||||||||||||||
| 5.2.3.3 | In case, if retesting is not done during laboratory investigations and proceeded further for full scale investigation; below procedure shall be followed. | |||||||||||||||||||||
| 5.2.3.4 | Retesting shall be performed by second analyst (Analyst II) with original sample in triplicate (3 test preparations). If the test results are individually within the specification performed by Analyst II, retesting of original portion of the sample in triplicate (3 test preparations) shall be performed by third analyst or original analyst (Analyst III or Analyst I) to confirm the test result obtained by analyst II. | |||||||||||||||||||||
| 5.2.3.5 | If all the retest results obtained are individually within the specification and %RSD of six results meet the acceptance criteria as described in table-1, all the retest results along with the average result shall be reported in the TDS. The report shall be submitted to QA in-charge for review and decision. | |||||||||||||||||||||
| 5.2.3.6 | The decision on the batch shall be taken after thorough review of investigation. Scientific justification for such decision shall be documented and approved by QA in-charge. In case of batch release, the average result shall substitute the initial test result and considered for reporting. | |||||||||||||||||||||
| 5.2.3.7 | If any of the test results is not within the specification limits and / or the % RSD of six results do not meet the acceptance criteria as described in table-1, the OOS results shall be treated as valid. Initial results shall be considered for reporting. Further decision for the disposition of the batch shall be taken by QA In-charge. | |||||||||||||||||||||
| 5.2.3.8 | If the retest results are individually within the specification limits and % RSD of six results do not meet the acceptance criteria as described in table-1, the OOS results shall be treated as valid. In such cases investigation shall be carried out to identify the cause for variability of the results. Based on the investigation outcome, further decision for the disposition of the batch shall be taken (as appropriate) by QA In-charge. | |||||||||||||||||||||
| 5.2.3.9 | The raw data of the results obtained on retesting shall be retained with original test data sheet. | |||||||||||||||||||||
| 5.3 | Procedure for Purchased Raw Material and Packaging Material: | |||||||||||||||||||||
| 5.3.1 | Follow stages described in step nos. 5.1.1 to 5.1.7, 5.1.11 to 5.1.12 and 5.1.14 to 5.1.16. | |||||||||||||||||||||
| 5.3.2 | If no laboratory error is identified then the OOS result is valid. The batch shall be rejected and dispositioned as appropriate. | |||||||||||||||||||||
| 5.3.3 | In case of packaging material, OOS investigation as per this SOP is applicable only in case of quantitative chemical tests and does not apply to test for text matter, colour, dimension checks etc. | |||||||||||||||||||||
| 5.4 | Procedure for OOS Test Results Obtained on Stability Sample: | |||||||||||||||||||||
| 5.4.1 | Follow stages described in the section 5.1.1 to 5.1.10, 5.1.12, 5.1.13 (for OOS noted in dissolution test), 5.1.16 and 5.1.17. | |||||||||||||||||||||
| 5.4.2 | In case of test batches, stability study may however be continued to generate additional data in discussion with PDL / R&D. | |||||||||||||||||||||
| 5.4.3 | For those drug product batches distributed in U.S.A., unless the OOS test results is found to be invalid, a Field Alert Report (FAR) shall be submitted to FDA within 3 working days of the OOS incident . A follow-up FAR shall be submitted when the OOS investigation has been completed. | |||||||||||||||||||||
| 5.4.4 | In case of products for European Market, information about OOS results shall be reported to Qualified Person in respective country as a part of Annual Product Review report. | |||||||||||||||||||||
| 5.4.5 | For test result occurred on commercial distributed batches for other than US and Europe, it shall be handled as per respective countries regulatory requirement. | |||||||||||||||||||||
| 5.4.6 | The OOS shall not be raised for the tests where the results are to be given against requirements of ‘For Information’ or ‘For Data Generation’. | |||||||||||||||||||||
| 5.5 | For OOS Test Results Obtained on Sample Tested by Contract / Public Testing Lab: | |||||||||||||||||||||
| 5.5.1 | Contract / Public testing laboratory shall notify any OOS result, This shall be defined in the agreement signed between plant and the Contract / Public testing laboratory. | |||||||||||||||||||||
| 5.5.2 | QA In-charge shall review the information received from the Contract / Public testing laboratory along with QC In-charge. | |||||||||||||||||||||
| 5.5.3 | If this review confirms the validity of OOS result, then phase – II investigation shall be initiated. In case retesting is necessitated as part of this investigation, this shall be done at the same Contract / Public testing laboratory. | |||||||||||||||||||||
| 5.6 | Impact Assessment | |||||||||||||||||||||
| 5.6.1 | In case where OOS is confirmed and assignable cause identified during investigation, assessment of impact on other manufactured batches of the product or different product (as applicable) shall be carried out by QA and documented. | |||||||||||||||||||||
| 5.7 | General Requirements For Handling Of OOS Results | |||||||||||||||||||||
| 5.7.1 | OOS investigation shall be completed and closed within 30 days of occurrence with an exception of 45 days for OOS of delay study analysis. In the event that an investigation cannot be completed within 30 days / 45 days for OOS of delay study analysis, an interim report shall be prepared. This shall contain summary of investigations carried out till date and further investigation plan and timeline for completion. QA In-charge shall confirm for the same and shall allow such extension only once, unless otherwise approved by Head-Quality in case of exceptions. | |||||||||||||||||||||
| 5.7.2 | Separate logs shall be maintained for OOS obtained for purchased raw materials / packaging materials; intermediates / finished product and stability samples. OOS log shall be updated on completion of the investigation. | |||||||||||||||||||||
| 5.7.3 | OOS result noted for different tests for a particular batch shall be logged and reported as separate OOS. | |||||||||||||||||||||
| 5.7.4 | The SOP shall not be applicable to the batches prepared for market samples, pre-purchase raw material and non-routine samples. | |||||||||||||||||||||
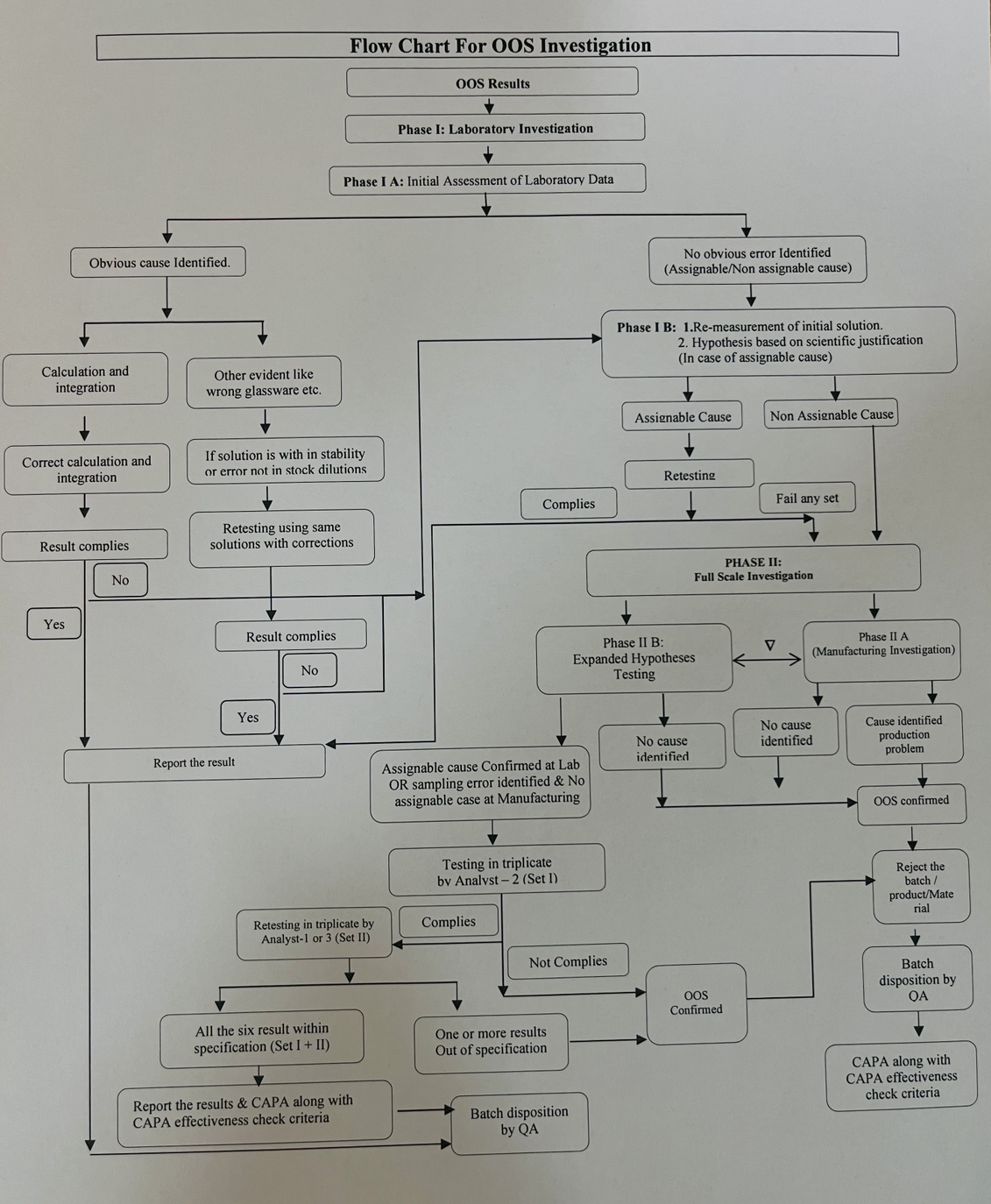
| 5.7.5 | The overall flow of OOS investigation and decision making tree of as given in Annexure 5. | |||||||||||||||||||||
| 5.7.6 | Review of OOS test results shall be done every six months. The review shall include trending of OOS test results based on the causes (eg.: analyst error, instrument error, method / specification error, equipment error, process error, human error in manufacturing, input material etc.). This classification shall be done product-wise / functional area wise as applicable. Trends shall be reviewed as a part of Annual Product Review and Management Review. Any emerging trends shall be evaluated for taking corrective actions and preventive actions. Trends generated in the previous year(s) shall be referred when evaluating trends for the current six month period. | |||||||||||||||||||||
| 5.7.7 | OOS test results records i.e., Log for OOS Test Results, OOS Investigation Form and trends shall be retained as per SOP on “Storage, Retrieval and Destruction of Records” | |||||||||||||||||||||
| 5.7.8 | In case of ‘Closed’ OOS needs to be re-opened for further investigation or impact assessment, then additional investigation report and / or necessary attachments shall be appended to the original report. Such re-opened OOS shall be closed within 30 days from the date of re-opening. This investigation report shall be attached with original investigation report as Addendum of Original OOS investigation. | |||||||||||||||||||||
| 5.7.9 | After complete investigation, if a batch is confirmed to be rejected; it shall be kept segregated and ‘Rejected’ label shall be pasted. | |||||||||||||||||||||