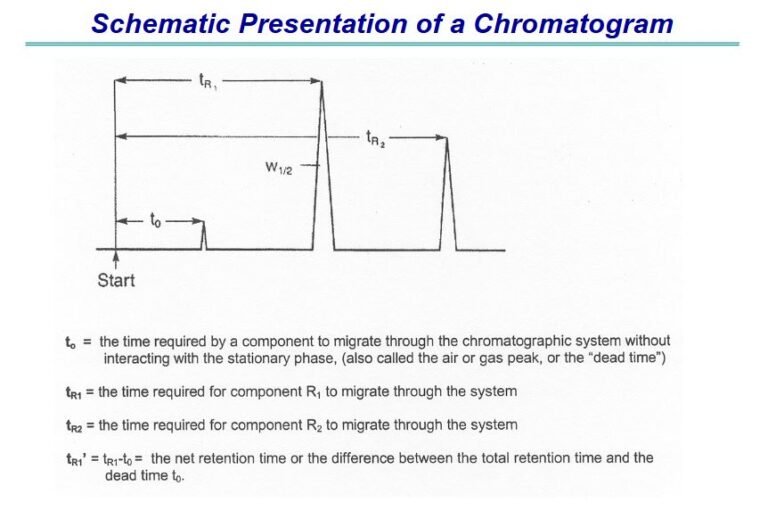
Few important Questions and Answer related to ANDA submission are as:–
- To which generic drug product submissions do this guidance and the RTR Standards guidance apply?
This guidance and the RTR Standards guidance apply to original abbreviated new drug applications (ANDAs) and certain prior approval supplements (PASs) to ANDAs consistent with our current practices, including those in which the applicant is seeking approval of a new strength11 , reformulation of a drug product that does not require a new original ANDA submission, return of a discontinued product to the market, and Rx-to OTC switches for all conditions of use.
- An applicant receives an IR listing minor deficiencies identified during the filing review. The IR states that the response is due within seven calendar days. What happens if the applicant fails to submit the responses within this time frame?
If an ANDA applicant receives an IR from FDA listing minor deficiencies identified during the filing review, and the requested information is not submitted and received within seven calendar days, FDA will RTR the ANDA. Responses to IRs should completely address all outstanding issues identified in the IR. Responses to IRs should be formatted and submitted following current electronic Common Technical Document (eCTD) specifications, submission structure recommendations, file format, and version recommendations (see guidance for industry on Providing Regulatory Submissions in Electronic Format – Certain Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications).
- An applicant receives an RTR determination and provides a response to some of the deficiencies. Will the response be accepted as a resubmission?
No, FDA will not accept a partial response to an RTR determination. If FDA issues an RTR determination, an applicant may either correct all deficiencies identified therein by resubmission of a complete ANDA (i.e., a new application that remedies the major deficiencies and any and all minor deficiencies identified in the RTR letter) or withdraw the application under 21 CFR 314.99. (If the applicant takes no action, FDA may consider the ANDA withdrawn after one year.)
- An applicant receives an RTR determination. The applicant resubmits the ANDA and provides a response to each deficiency, but the applicant receives a subsequent RTR determination. Why did FDA RTR the resubmission?
FDA may RTR a resubmission for multiple reasons including, but not limited to, the failure to provide a comprehensive response to the deficiencies identified in the RTR letter and failure to follow the current recommendations relevant to filing (e.g., recommendations set forth in RTR guidance’s and in product-specific bioequivalence guidance’s in effect at the time of resubmission.
- How long does an applicant have to request reconsideration of an RTR determination?
The Agency believes that seven calendar days provides an applicant sufficient time to review FDA’s regulatory action and determine whether the applicant would like to pursue a request for reconsideration. It also ensures that applicants submit requests for reconsideration of recent Agency regulatory actions. Like minor deficiencies, any matters that will be challenged should be reviewed, analyzed, and addressed within seven calendar days.
The Agency recognizes that an applicant will require some time to identify, coordinate, and compile the information necessary to submit a response to the RTR determination; however, in an effort to streamline reviews and enable timely inspections, the Agency needs to define an acceptable time frame in which to challenge an RTR determination. For example, if an applicant receives an RTR determination 60 days after the application is submitted and submits a successful request for reconsideration one month after the RTR determination was made, the Agency will have lost one month of application review time and the Agency’s ability to ensure timely inspections may be jeopardized
- Will FDA accept an unsolicited filing amendment submitted during filing review to correct a deficiency identified by the ANDA applicant?
FDA will not review any unsolicited amendment submitted by the applicant, other than an administrative amendment identifying a change in contact information or ownership, while the ANDA is pending filing review. For example, an amendment containing data that should have been included in the original submission will not be reviewed during the filing review or considered when making the determination of whether the ANDA is a substantially complete application.
- If a typographical error (or “typo”) is the basis for a major deficiency resulting in an RTR determination, will FDA rescind its RTR decision if the applicant identifies the typographical error and provides a corrected submission?
FDA will not rescind the RTR determination if a typographical error is the basis for a major deficiency and the applicant informs FDA of the error and provides a corrected submission. When compiling an ANDA, applicants should ensure that all relevant data and information necessary to support the substantive review is correctly transcribed into appropriate sections of the ANDA. When performing a filing review, the Agency will assume that the information transcribed in an ANDA is correct. The Agency must move expeditiously through the filing process to ensure that FDA is able to meet its commitment to review and act on ANDAs within specified GDUFA performance metric goal dates.
A typographical error that may result in a major deficiency due to an applicant’s lack of quality control is not limited to a numerical value; the error may involve an alphabetical letter, symbol, or other text.
Example: In guidance, FDA recommends that ANDAs contain the initiation date for each stability study conducted, along with individual pull dates (removal from the storage chamber and preferably identified in MM/DD/YYYY or YYYY/MM/DD format) for each stability time point, so FDA can verify that each study covers the recommended six-month (180-day) minimum hold time. If the applicant provides a date that does not confirm a 180-day hold time as a result of a typographical error, FDA will RTR the ANDA. A demonstration by the applicant that the basis of the RTR was caused by a typographical error will not be sufficient to rescind the RTR determination for this type of deficiency.
Example: In guidance, FDA recommends that ANDAs contain information concerning Long-Term Storage Stability coverage in the applicable row of the Study Information Bioequivalence table. These data are necessary to ascertain adherence to appropriate storage temperatures and duration. If the applicant provides a date or temperature (including temperature range) that does not confirm proper storage conditions or duration as a result of a typographical error, FDA will RTR the ANDA. A demonstration by the applicant that the basis of the RTR was caused by a typographical error will not be sufficient to rescind the RTR determination for this type of deficiency.
Example: In guidance, FDA recommends that a threshold value for a particular variable should be >60 units. In the ANDA, the applicant uses a symbol that indicates it does not meet the threshold value (e.g., <60 units) As a result of this typographical error, FDA will RTR the ANDA. A demonstration by the applicant that the basis of the RTR was caused by a typographical error will not be sufficient to rescind the RTR determination for this type of deficiency.
- Will FDA RTR an ANDA that does not contain a patent certification that is 256 consistent with the regulations?
FDA will treat as a minor deficiency an applicant’s failure to include a patent 259 certification or statement that is consistent with section 505(j)(2)(A)(vii) and (viii) of the 260 FD&C Act and 21 CFR 314.94(a)(12) and 314.96(d).
- Will FDA RTR an ANDA if the submission contains a section or information in a 263 language other than English and the applicant does not provide an English 264 translation?
FDA will identify untranslated content, which must be translated into English, in an IR. Failure to provide the requested translations will result in an RTR. Moreover, it is incumbent upon the applicant to ensure that any and all untranslated content in the ANDA submission, including all sections of the document (e.g., headers, titles), is translated into English. Therefore, should FDA discover additional untranslated content in the ANDA submission after an IR response has been submitted, the ANDA will be refused for receipt.
FDA will accept an ANDA with the English translation on a page next to the original text. FDA recommends that the translation be printed in size 12 type to facilitate review. The applicant should use its best judgment in determining how to fit the necessary information on a page without impacting the reviewer’s ability to read the information.
- Will FDA RTR an ANDA that fails to use two API lots to manufacture three batches 350 of each strength of a proposed product?
Yes, FDA will consider an applicant’s failure to use two API lots to manufacture three batches of each strength of a proposed product a major deficiency.
- Will FDA RTR an ANDA if the proposed packaging is inconsistent with the condition(s) of use approved for the reference listed drug (RLD)?
Yes, FDA will RTR an ANDA if the proposed packaging is inconsistent with the condition(s) of use approved for the RLD. For example, if the RLD is approved for a 14-day course of treatment, repeated at a specific interval, and is marketed in 14-count packages, the ANDA should propose marketing containers that are consistent with the approved condition(s) of use.
- Will FDA RTR an ANDA that references batch records previously submitted by an 400 applicant in another ANDA?
Yes, FDA will RTR an ANDA that references batch records previously submitted by an applicant in another ANDA. When an applicant submits individual ANDAs for drug products containing the same active ingredient, each ANDA should have its own independent basis for approval.
- The guidance for industry ANDA Submissions – Refuse to Receive for Lack of Justification of Impurity Limits indicates that FDA will RTR an ANDA that fails to provide justification for certain impurities above specified thresholds. How does FDA review ANDAs at filing to determine if sufficient justification is provided in the application?
During the filing review, FDA will determine if the ANDA: 1) proposes limits in drug substances and drug products for specified identified impurities that are above qualification thresholds; (2) proposes limits for specified unidentified impurities that are above identification thresholds; and/or (3) proposes limits for unspecified impurities (e.g., any unknown impurity) that are above identification thresholds. If any of these factors are met, FDA will review the ANDA to determine if the applicant has provided a justification for these proposals. If the ANDA does contain a justification, the application will be received, assuming there are no deficiencies leading to an RTR determination. The sufficiency of the justification will be reviewed during the technical review of the ANDA. If the ANDA does not contain a justification for the proposed limits, FDA will RTR the application.
- What is the proper location for comparative (test product and RLD) half-tablet dissolution data?
FDA recommended half-tablet dissolution test data should be contained in Module 2.7 of the ANDA. The failure to perform the recommended half-tablet dissolution studies (i.e., test product and RLD) or the failure to correctly place such dissolution data in Module 2.7 is considered a major deficiency.
For more details refer: ANDA Submissions – Refuse-to-Receive Standards: Questions and Answers Guidance for Industry.