Chromatographic separation techniques are multistage separation procedures in which the components of a sample are distributed between two phases, one of which is stationary while the other is mobile. The stationary phase may be a solid or a liquid supported on a solid or a gel. The stationary phase may be packed in a column, spread as a layer, or distributed as a film, etc. The mobile phase may be gaseous or liquid or supercritical fluid. The separation may be based on adsorption, mass distribution (partition), ion exchange, etc., or may be based on differences in the physicochemical properties of the molecules such as size, mass, volume, etc.
Portions of the present general chapter text that are national USP–NF text, and therefore not part of the harmonized text, are marked with symbols (♦♦) to specify this fact.
♦This chapter describes general procedures, definitions, and calculations of common parameters and generally applicable requirements for system suitability.
The types of chromatography useful in qualitative and quantitative analysis employed in USP procedures are column, gas, paper, thin-layer (including high-performance thin-layer chromatography), and pressurized liquid chromatography (commonly called high-pressure or high-performance liquid chromatography).
GENERAL PROCEDURES
This section describes the basic procedures used when a chromatographic method is described in a monograph. The following procedures are followed unless otherwise indicated in the individual monograph.
Paper Chromatography
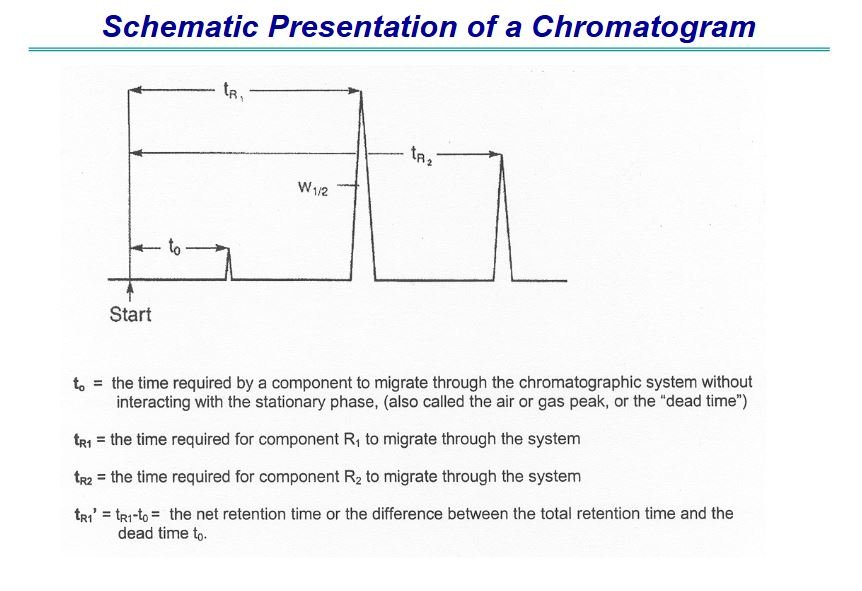
stationary phase
The stationary phase is a sheet of paper of suitable texture and thickness. Development may be ascending, in which the solvent is carried up the paper by capillary forces, or descending, in which the solvent flow is also assisted by gravitational force. The orientation of paper grain with respect to solvent flow is to be kept constant in a series of chromatograms. (The machine direction is usually designated by the manufacturer.)
apparatus
The essential equipment for paper chromatography consists of a vapor-tight chamber with inlets for addition of solvent and a rack of corrosion-resistant material about 5 cm shorter than the inside height of the chamber. The rack serves as a support for solvent troughs and for antisiphon rods that, in turn, hold up the chromatographic sheets. The bottom of the chamber is covered with the prescribed solvent system or mobile phase. Saturation of the chamber with solvent vapor is facilitated by lining the inside walls with paper wetted with the prescribed solvent system.
spotting
The substance or substances analyzed are dissolved in a suitable solvent. Convenient volumes delivered from suitable micropipets of the resulting solution, normally containing 1–20 µg of the compound, are placed in 6- to 10-mm spots not less than 3 cm apart.
descending paper chromatography procedure
A spotted chromatographic sheet is suspended in the apparatus, using the antisiphon rod to hold the upper end of the sheet in the solvent trough. [Note—Ensure that the portion of the sheet hanging below the rods is freely suspended in the chamber without touching the rack, the chamber walls, or the fluid in the chamber.]
The chamber is sealed to allow equilibration (saturation) of the chamber and the paper with the solvent vapor. Any excess pressure is released as necessary.
After equilibration of the chamber, the prepared mobile phase is introduced into the trough through the inlet.
The inlet is closed, and the mobile solvent phase is allowed to travel the desired distance down the paper.
The sheet is removed from the chamber.
The location of the solvent front is quickly marked, and the sheet is dried.
The chromatogram is observed and measured directly or after suitable development to reveal the location of the spots of the isolated drug or drugs.
ascending paper chromatography procedure
The mobile phase is added to the bottom of the chamber.
The chamber is sealed to allow equilibration (saturation) of the chamber and the paper with the solvent vapor. Any excess pressure is released as necessary.
The lower edge of the stationary phase is dipped into the mobile phase to permit the mobile phase to rise on the chromatographic sheet by capillary action.
When the solvent front has reached the desired height, the chamber is opened, the sheet is removed, the location of the solvent front is quickly marked, and the sheet is dried.
The chromatogram is observed and measured directly or after suitable development to reveal the location of the spots of the isolated drug or drugs.
Thin-Layer Chromatography
stationary phase
The stationary phase is a relatively thin, uniform layer of dry, finely powdered material applied to a glass, plastic, or metal sheet or plate (typically called the plate). The stationary phase of thin-layer chromatography (TLC) plates has an average particle size of 10–15 µm, and that of high-performance TLC (HPTLC) plates has an average particle size of 5 µm. Commercial plates with a preadsorbent zone can be used if they are specified in a monograph. Sample applied to the preadsorbent region develops into sharp, narrow bands at the preadsorbent–sorbent interface. The separations achieved may be based on adsorption, partition, or a combination of both effects, depending on the particular type of stationary phase.
apparatus
A chromatographic chamber made of inert, transparent material and having the following specifications is used: a flat-bottom or twin trough, a tightly fitted lid, and a size suitable for the plates. The chamber is lined on at least one wall with filter paper. Sufficient mobile phase or developing solvent is added to the chamber so that, after impregnation of the filter paper, a depth appropriate to the dimensions of the plate used is available. The chromatographic chamber is closed and allowed to equilibrate. [Note—Unless otherwise indicated, the chromatographic separations are performed in a saturated chamber.]
detection/visualization
An ultraviolet (UV) light source suitable for observations under short- (254 nm) and long- (365 nm) wavelength UV light and a variety of spray reagents to make spots visible are often used.
spotting
Solutions are spotted on the surface of the stationary phase (plate) at the prescribed volume in sufficiently small portions to obtain circular spots of 2–5 mm in diameter (1–2 mm on HPTLC plates) or bands of 10–20 mm × 1–2 mm (5–10 mm × 0.5–1 mm on HPTLC plates) at an appropriate distance from the lower edge and sides of the plate. [Note—During development, the application position must be at least 5 mm (TLC) or 3 mm (HPTLC) above the level of the mobile phase.] The solutions are applied on a line parallel to the lower edge of the plate with an interval of at least 10 mm (5 mm on HPTLC plates) between the centers of spots, or 4 mm (2 mm on HPTLC plates) between the edges of bands, then allowed to dry.
procedure
Place the plate in the chamber, ensuring that the spots or bands are above the surface of the mobile phase.
Close the chamber.
Allow the mobile phase to ascend the plate until the solvent front has traveled three-quarters of the length of the plate, or the distance prescribed in the monograph.
Remove the plate, mark the solvent front with a pencil, and allow to dry.
Visualize the chromatograms as prescribed.
Determine the chromatographic retardation factor (RF) values for the principal spots or zones.
Presumptive identification can be made by observation of spots or zones of identical RF value and about equal magnitude obtained, respectively, with an unknown and a standard chromatographed on the same plate. A visual comparison of the size or intensity of the spots or zones may serve for semiquantitative estimation. Quantitative measurements are possible by means of densitometry (absorbance or fluorescence measurements).
Column Chromatography
solid support
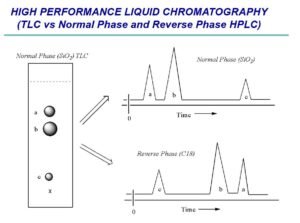
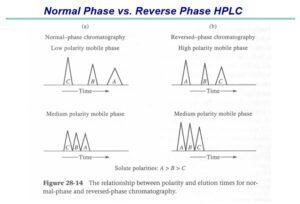
Purified siliceous earth is used for normal-phase separation. Silanized chromatographic siliceous earth is used for reverse-phase partition chromatography.
stationary phase
The solid support is modified by the addition of a stationary phase specified in the individual monograph. If a mixture of liquids is used as the stationary phase, mix the liquids before the introduction of the solid support.
mobile phase
The mobile phase is specified in the individual monograph. If the stationary phase is an aqueous solution, equilibrate with water. If the stationary phase is a polar organic fluid, equilibrate with that fluid.
apparatus
Unless otherwise specified in the individual monograph, the chromatographic tube is about 22 mm in inside diameter and 200–300 mm in length. Attached to it is a delivery tube, without stopcock, about 4 mm in inside diameter and about 50 mm in length.
Apparatus preparation:
Pack a pledget of fine glass wool in the base of the tube. Combine the specified volume of stationary phase and the specified amount of solid support to produce a homogeneous, fluffy mixture. Transfer this mixture to the chromatographic tube, and tamp using gentle pressure to obtain a uniform mass. If the specified amount of solid support is more than 3 g, transfer the mixture to the column in portions of approximately 2 g, and tamp each portion. If the assay or test requires a multisegment column with a different stationary phase specified for each segment, tamp after the addition of each segment, and add each succeeding segment directly to the previous one. Pack a pledget of fine glass wool above the completed column packing. [Note—The mobile phase should flow through a properly packed column as a moderate stream or, if reverse-phase chromatography is applied, as a slow trickle.]
If a solution of the analyte is incorporated into the stationary phase, complete the quantitative transfer to the chromatographic tube by scrubbing the beaker used for the preparation of the test mixture with a mixture of about 1 g of solid support and several drops of the solvent used to prepare the sample solution before adding the final portion of glass wool.
procedure
Transfer the mobile phase to the column space above the column packing, and allow it to flow through the column under the influence of gravity.
Rinse the tip of the chromatographic column with about 1 mL of mobile phase before each change in composition of mobile phase and after completion of the elution.
If the analyte is introduced into the column as a solution in the mobile phase, allow it to pass completely into the column packing, then add mobile phase in several small portions, allowing each to drain completely, before adding the bulk of the mobile phase.
Where the procedure indicates the use of multiple chromatographic columns mounted in series and the addition of mobile phase in divided portions is specified, allow each portion to drain completely through each column, and rinse the tip of each with mobile phase before the addition of each succeeding portion.
Gas Chromatography (GC)
liquid stationary phase
This type of phase is available in packed or capillary columns.
packed column GC
The liquid stationary phase is deposited on a finely divided, inert solid support, such as diatomaceous earth, porous polymer, or graphitized carbon, which is packed into a column that is typically 2–4 mm in internal diameter and 1–3 m in length.
capillary column GC
In capillary columns, which contain no packed solid support, the liquid stationary phase is deposited on the inner surface of the column and may be chemically bonded to it.
solid stationary phase
This type of phase is available only in packed columns. In these columns the solid phase is an active adsorbent, such as alumina, silica, or carbon, packed into a column. Polyaromatic porous resins, which are sometimes used in packed columns, are not coated with a liquid phase. [Note—Packed and capillary columns must be conditioned before use until the baseline and other characteristics are stable. The column or packing material supplier provides instructions for the recommended conditioning procedure.]
apparatus
A gas chromatograph consists of a carrier gas source, injection port, column, detector, and recording device. The injection port, column, and detector are temperature controlled and may be varied as part of the analysis. The typical carrier gas is helium, nitrogen, or hydrogen, depending on the column and detector in use. The type of detector used depends on the nature of the compounds analyzed and is specified in the individual monograph. Detector output is recorded as a function of time, and the instrument response, measured as peak area or peak height, is a function of the amount present.
temperature program
The length and quality of a GC separation can be controlled by altering the temperature of the chromatographic column. When a temperature program is necessary, the individual monograph indicates the conditions in table format. The table indicates the initial temperature, rate of temperature change (ramp), final temperature, and hold time at the final temperature.
procedure
Equilibrate the column, injector, and detector with flowing carrier gas until a constant signal is received.
Inject a sample through the injector septum, or use an autosampler.
Begin the temperature program.
Record the chromatogram.
Analyze as indicated in the monograph.
Liquid Chromatography
The term “liquid chromatography” (LC), as used in the compendia, is synonymous with high-pressure liquid chromatography and high-performance liquid chromatography. LC is a separation technique based on a solid stationary phase and a liquid mobile phase.
stationary phase
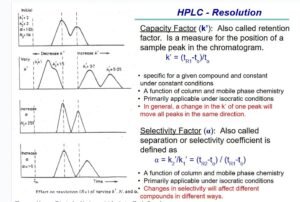
Separations are achieved by partition, adsorption, or ion-exchange processes, depending on the type of stationary phase used. The most commonly used stationary phases are modified silica or polymeric beads. The beads are modified by the addition of long-chain hydrocarbons. The specific type of packing needed to complete an analysis is indicated by the “L” designation in the individual monograph (see also the section Chromatographic Columns). The size of the beads is often described in the monograph as well. Changes in the packing type and size are covered in the System Suitability section of this chapter.
chromatographic column
The term “column” includes stainless steel, lined stainless steel, and polymeric columns, packed with a stationary phase. The length and inner diameter of the column affects the separation, and therefore typical column dimensions are included in the individual monograph. Changes to column dimensions are discussed in the System Suitability section of this chapter. Compendial monographs do not include the name of appropriate columns; this omission avoids the appearance of endorsement of a vendor’s product and natural changes in the marketplace. See the section Chromatographic Columns for more information.
In LC procedures, a guard column may be used with the following requirements, unless otherwise is indicated in the individual monograph: (a) the length of the guard column must be NMT 15% of the length of the analytical column, (b) the inner diameter must be the same or smaller than that of the analytical column, and (c) the packing material should be the same as the analytical column (e.g., silica) and contain the same bonded phase (e.g., C18). In any case, all system suitability requirements specified in the official procedure must be met with the guard column installed.
mobile phase
The mobile phase is a solvent or a mixture of solvents, as defined in the individual monograph.
apparatus
A liquid chromatograph consists of a reservoir containing the mobile phase, a pump to force the mobile phase through the system at high pressure, an injector to introduce the sample into the mobile phase, a chromatographic column, a detector, and a data collection device.
gradient elution
The technique of continuously changing the solvent composition during the chromatographic run is called gradient elution or solvent programming. The gradient elution profile is presented in the individual monograph as a gradient table, which lists the time and proportional composition of the mobile phase at the stated time.
procedure
Equilibrate the column and detector with mobile phase at the specified flow rate until a constant signal is received.
Inject a sample through the injector, or use an autosampler.
"A seasoned professional with a Master's in Chemistry, the author brings more than two decades of expertise in Quality Control, Analytical Method Validation, and advanced QMS investigation. Their experience is globally validated by successful management of major regulatory audits, including those conducted by the US, EU, Brazil, and Russian authorities."